General JavaProtein Dossier Area Help
Java Protein
Dossier is an interactive presentation of important physical-chemical
characteristics of the macromolecular structure described in PDB file.
With a few mouse clicks a user can access data about chosen parameter,
call other BLUE STAR STING modules or refine the search for a specific characteristic.
By using color code scales for each residue of the sequence, JPD shows
corresponding: temperature factor, solvent accessibility of the single
chain (and also in complex with the other present chains in given PDB
file), hydrophobicity, sequence conservation in a multiple alignment (relative
entropy), double occupancies, reliability and histograms representing
the atomic contacts. JPD also shows the identification of Interface Forming
Residue (IFR) and their internal contacts. JPD offers information about
electrostatic potential and curvature on protein surface. In addition,
comparison of the Secondary Structure annotated by PDB, by DSSP and by
Stride is presented. The JPD_HELP below is presented in the separate organizational
units, so that a user is capable to quickly understand what is available
in JPD DataBase and how to access this information, in addition to instructions
which will show to a user what type of output he/she should expect. We
relay much on the image summaries, rather than using the words.
INDEX
| JPD Select HELP | References | Energy values for different contact types |
| Handling Local Files | ||
This is a general area at the java window of JPD (click on any of the
titles and you will get into more detailed explanation about highlighted
areas):
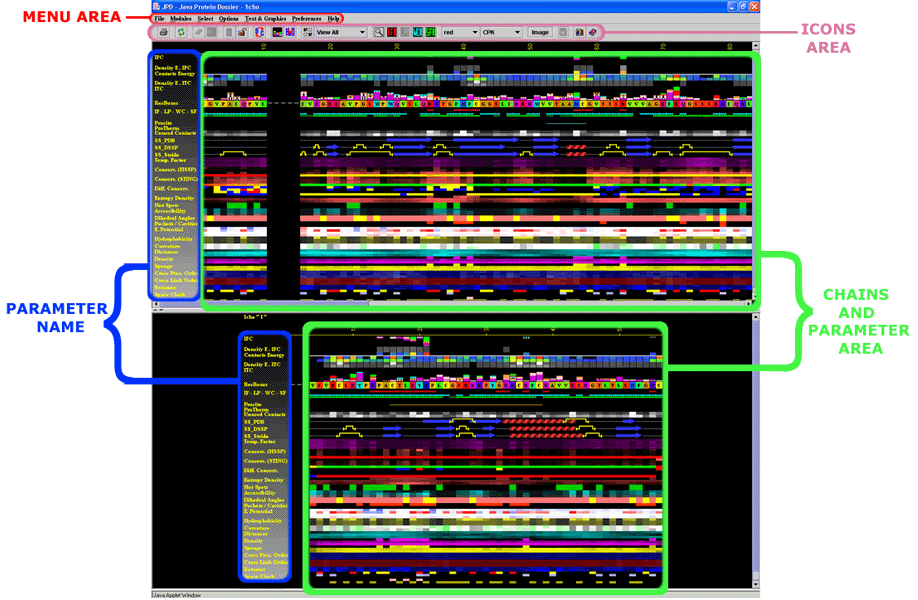
 |
IFC:
InterFace
Residue area Contacts - interatomic contacts
established between residues belonging to two different chains facing
each other. |
| Density
E.IFC: Energy
Density of the IFC
- The sum of Energies (calculated according to the table
of energy values for each contact type) for the contacts
established within a given sphere, among the residues belonging to
two different chains facing each other, is calculated and then divided
by the volume of the sphere. |
|
| Contacts
Energy
- Sum of the Energies of contacts established among residues belonging
to the same protein chain. (See the table
of energy values for each contact type) |
|
| Density
E.ITC: Energy
Density of ITC
- The sum of Energies (calculated according to the table
of energy values for each contact type) for the contacts
established within a given sphere, among residues belonging to the
same protein chain , is calculated and then divided by the volume
of the sphere. |
|
| ITC:
Internal Contacts -
Interatomic contacts among residues belonging to the same protein
chain. |
|
| ResBoxes: Residue Boxes - A single letter code is shown, representing the amino acids of the protein sequence which structure is inspected. The amino acid Boxes are color coded according to either STING_Paint code (1) or according to William Taylor (2) code. | |
| IF:
Interface
area - Residues identified at the interface between two protein chains. LP: Ligand Pocket Residues WC: Internal, protein co-crystalized Water Contacting Residues SF: SurFace residues (having contact with a solvent) |
|
| Prosite: Prosite pattern identification (3) | |
| Unused
Contacts
- Each residue can make certain (max)
number of contacts. The difference between the max number
of contacts and the contacts established, is presented here.
|
|
| SS_PDB:
Secondary Structure according to PDB
(4) file annotation. SS_DSSP: Secondary Structure according to DSSP(5) annotation. SS_STRIDE: Secondary Structure according to STRIDE (6) annotation. |
|
| Mult. Occupancy -A presence of two or more sets of coordinates for the same atom/residue in the PDB file is due to the electron density map interpretation where the experiment registered a diffraction from the crystals freezing the same molecule but with the different space positions for a certain amino acid. | |
| Temp. Factor: The temperature factor as annotated in a PDB file is presented. | |
| Conserv.
(HSSP):
The Amino acid sequence conservation and reliability
according to HSSP (7)
data is presented here. The Evolutionary Pressure, calculated based
on HSSP alignments, adequately prepared by BLUE STAR STING to be served
as an input to Rate4Site (8) software,
is also shown.
|
|
| Conserv.(STING)
The
Amino acid sequence conservation and reliability according to SH2Qs
data is shown here. The Evolutionary Pressure, calculated based on
SH2Qs alignments (adequately prepared by BLUE STAR
STING) to be served as an input to Rate4Site
(8) software, is also presented.
|
|
| Diff. Conserv. - The Difference in relative entropy, reliability and evolutionary pressure for HSSP and SH2Qs alignments is shown | |
| Entropy Density - The sum of values is calculated for the relative entropy (according to HSSP (7) data) of the amino acids encountered within the sphere of a given radius, and then divided by the volume of that sphere. | |
| Hot-Spots: This parameter indicates the existence of hydrophobic patches (9) at the surfaces of proteins. | |
| Accessibility:
The Amino acid accessibility is calculated according
to SurfV (10) program.
JPD shows 3 values: for the protein chain in isolation, for the protein
chain in complex with the other chain (if) present in the PDB file
and finally, a relative accessibility (the last one given by the table
of absolute solvent accessible area for amino acids).
|
|
| Dihedral
Angles - Dihedral angles are
calculated according to the original work by Ramashandran
(11) .
|
|
| Pockets/Cavities:
Pockets/cavities are calculated using using two different algorithms: Pocket, which is part of the package ProShape; and NanoShaper.
|
|
| E.
Potential
-
Electrostatic Potential is calculated using Delphi
(12) program according to the modifications done by Walter
Rocchia (13) and further adapted to JPD requirements (to
be published) |
|
| Hydrophobicity- The Hydrophobicity values are mapped according to the table with hydrophobicity values for 20 amino acids. | |
| Curvature
- The curvature values for each amino acid are calculated using the
program SurfeRace (14).
| |
| Distance
- The Distance from the N-terminal amino acid Ca atom, C-terminal
amino acid Ca atom and center of the protein mass point, is calculated
from any given amino acid starting from its Ca atom.
|
|
| Density
- The Density is calculated by the summation of atom mass for all
atoms encountered within a sphere of a given radius (centered either
at the CA [alpha carbon] or LHA [Last Heavy Atom] in the side chain
of this residue), and then dividing it by the volume of the sphere.
|
|
| Sponge
- Sponge is not an inverse of the Density! The Sponge is calculated
by the summation of van der waals volumes for all atoms encountered
within a sphere of a given radius (centered either at the CA [alpha
carbon] or LHA [Last Heavy Atom] in the side chain for this residue),
and then dividing it by the volume of the sphere.
|
|
| Order
of Cross Link
-The order of cross link is identified as a number of cross-links established
among independent stretches of sequence (the size of which varies
from 15, 20 to 30 Amino Acids). Cross Links are defined as contacts
(any type from possible 5 classes: 1. Hydrophobic interaction, 2.
Hydrogen Bonding, 3. Aromatic Stacking, 4. Salt bridging, 5. Cystein-bridging)
established among residues that are far apart in the protein primary
sequence, but are close in its 3D fold.
|
|
| Order
of Cross Presence
- Cross Presence is defined as presence within
a probing sphere (centered at a given residue) of any residue that
is far apart in the protein primary sequence from the central residue,
but is close in its 3D fold. The order is identified as a number of
such cross-presence encounters among independent stretches of sequence
(the size of which varies from 15, 20 to 30 Amino Acids).
|
|
| Space
Clash (5 categories)
- The steric clash
occurring among amino acids is reported here, measured by the "overlap
factor", which is defined as the ratio of the distance between
two atom centers to the sum of their van der Waals radii.
|
|
| ProTherm - ProTherm is a collection of numerical data of thermodynamic parameters such as Gibbs free energy change, enthalpy change, heat capacity change, transition temperature etc. for wild type and mutant proteins, that are important for understanding the structure and stability of proteins. | |
| Rotamers
- We have used the Rotamer Library presented in Lovell
et. Al. to calculate and then evaluate how rare is the rotamer configuration
for each residue in a given PDB file. |
|
| Color palette adjustment - Although the color scale used to represent each parameter is fixed (e.g., gray scale, red to blue, red to green), the values associated with the beginning and the end of the color scale for each parameter can be changed. This is done to facilitate the visualization of the variation of the parameter in the residues sequence. |
Generally, STING operates with both PDB public files and local files
in pdb format. However, in order to handle properly STING_DB parameters
for a local file, STING needs to pre-calculate those. Similarly, JPD can
handle both public and local files and this can be done for a single structure
and for two structurally aligned structures;
1. For the single structure, a user may submit
a job to the STING Server and obtain in his/her e-mail box a msg with
attached TGZ file, containing
all parameters that the JPD can display and analyze.
2. For two structurally aligned files JPD can handle both two publicly available (PDB deposited) files or two local (non public) pdb formatted files. In this case a user needs to submit two local files to the STING server and obtain two TGZ files before doing structural alignment. See more details here.
1. Neshich, G., Togawa, R., Mancini, A. L., Kuser, P. R., Yamagishi, M. E. B., Pappas Jr., G., Torres, W. V., Campos, T. F., Ferreira, L. L., Luna, F. M., Oliveira, A. G., Miura, R. T., Inoue, M. K., Horita, L. G., de Souza, D. F., Dominiquini, F., Álvaro, A., Lima, C. S., Ogawa, F. O., Gomes, B. G., Palandrani, J. C. F., dos Santos, G. F., de Freitas, E. M., Mattiuz, A. R., Costa, I. C., de Almeida, C. L., Souza, S., Baudet, C. and Higa, R. H. (2003) STING Millennium: a Web based suite of programs for comprehensive and simultaneous analysis of protein structure and sequence. Nucleic Acids Research, 31:13, 3386-3392.
2. W.R. Taylor (1997) "Residual colors: a proposal for aminochromography". Protein Eng, Jul;10(7):743-746.
3. Falquet L., Pagni M., Bucher P., Hulo N., Sigrist C.J., Hofmann K., Bairoch A.(2002) The PROSITE database, its status in 2002 Nucleic Acids Res. 30:235-238.
4. Berman, H.H., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N. and Bourne, P.E. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235-242.
5. Sander, C. and Schneider, R. (1991) Database of Homology-Derived Protein Structures and the Structural Meaning of Sequence Alignment. Proteins: Struc., Func. and Genet., 9, 56-68.
6. Frishman, D. and Argos, P. (1995) Knowledge-Based Protein Secondary Structure Assignment. Proteins: Struc., Func., and Genet., 23, 566-679.
7.Schneider, R and Sander, C. (1996) The HSSP database of protein structure-sequence alignments. Nucleic Acids Res., 24, 201-205.
8. Pupko T., Bell R.E., Mayrose I., Glaser F. and Ben-Tal N. Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics (2002) Jul;18 Suppl 1:S71-7.
9. Philip Lijnzaad, Herman J.C. Berendsen and Patrick Argos.(1996) A method for detecting hydrophobic patches on protein surfaces. Proteins: Structure, Function and Genetics 26: 192-203.
10. Sridharan, S., Nicholls, A. and Honig, B. (1992) A new vertex algorithm to calculate solvent accessible surface areas. Biophys. J., 61, A174.
11. Ramachandran, G. N., Ramakrishnan, C. and Sasisekharan, V. (1963) Stereochemistry of polypeptide chain configurations. J. Mol. Biol., 7, 95-99.
12. B. Honig and A. Nicholls, (1995) Classical electrostatics in biology and chemistry. Science 268 1144-1149 .
13. W. Rocchia, S. Sridharan, A. Nicholls, E. Alexov, A. Chiabrera and B. Honig, (2002) Rapid Grid-based Construction of the Molecular Surface for both Molecules and Geometric Objects: Applications to the Finite Difference Poisson-Boltzmann Method, J. Comp. Chem. 23:128-137
14. Tsodikov OV, Record MT Jr, Sergeev YV. ( 2002) Novel computer program for fast exact calculation of accessible and molecular surface areas and average surface curvature.J Comput Chem. Apr 30;23(6):600-9.
15. Radzicka, A. & Wolfenden, R. (1988). Comparing the polarities of the amino-acids -- side-chain distribution coefficients between the vapor-phase, cyclohexane, 1-octanol, and neutral aqueous-solution. Biochemistry 27, 1664-1670.