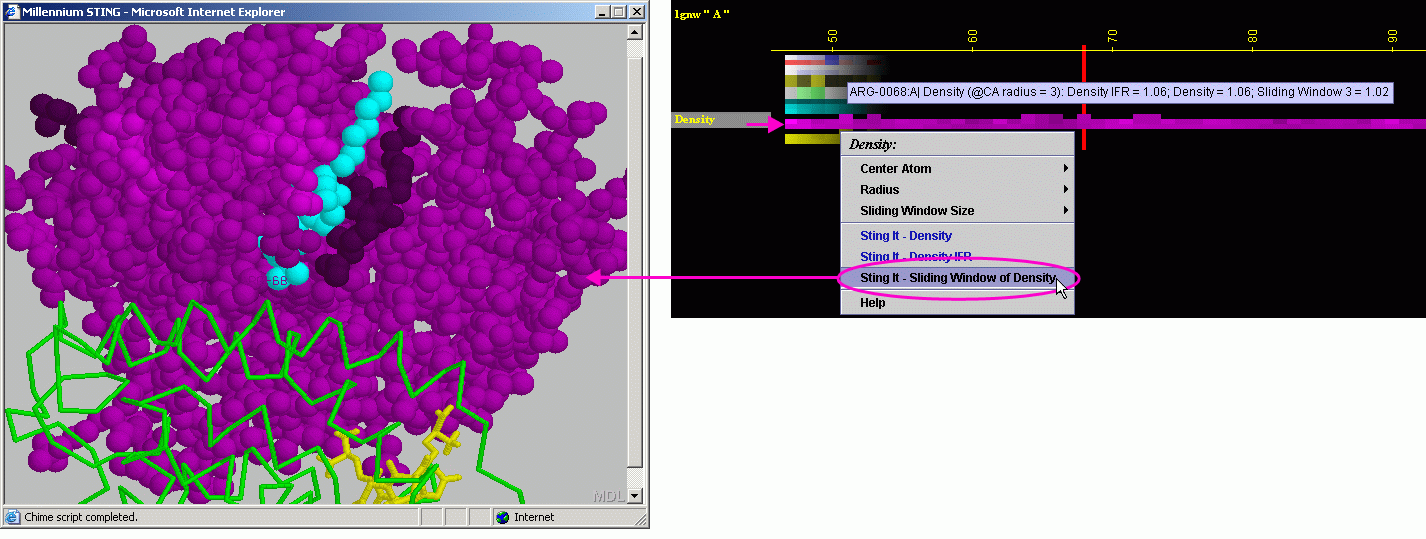
Density
The Density is calculated by the summation of atom mass for all atoms encountered within a sphere of a given radius (centered either at the CA [alpha carbon] or LHA [Last Heavy Atom] in the side chain of this residue), and then dividing it by the volume of the sphere.
Placing
the cursor above this element: pop-up area will show the position
(sequence number and AA three letter code) for selected amino acid and
the numerical values for Density (at interface (IFR), internal and sliding
window of given size) for this amino acid.
Left mouse click: no action
Right mouse
click: on any of the "Density" will generate following menu
and actions:
A) A user may select either
Ca or Last Heavy Atom (LHA) in the side chain as a center of the sphere
for calculating "Density"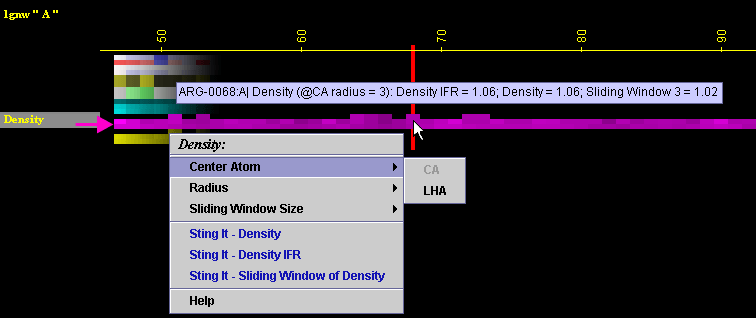
B) A user may select among
5 possible radius values for the sphere:
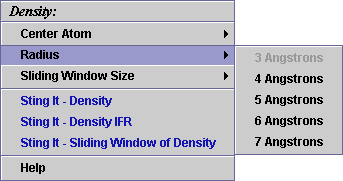
C) A user may select among
4 possible values for the sliding window size (for the size of 5: a value
for this parameter is collected over the central residue and other 2 from
the each side of the central one, summed and divided by 5):
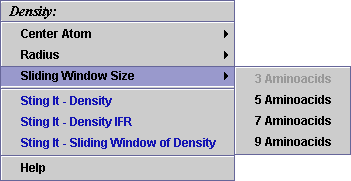
D) STING it "Density":
This option generates structural presentation with color coding of the
amino acids corresponding to the JPD row "Density" color coding.
The area of gaps is where the colors are going to be different from the
rest of the protein. Amino acids are presented in CPK rendering.
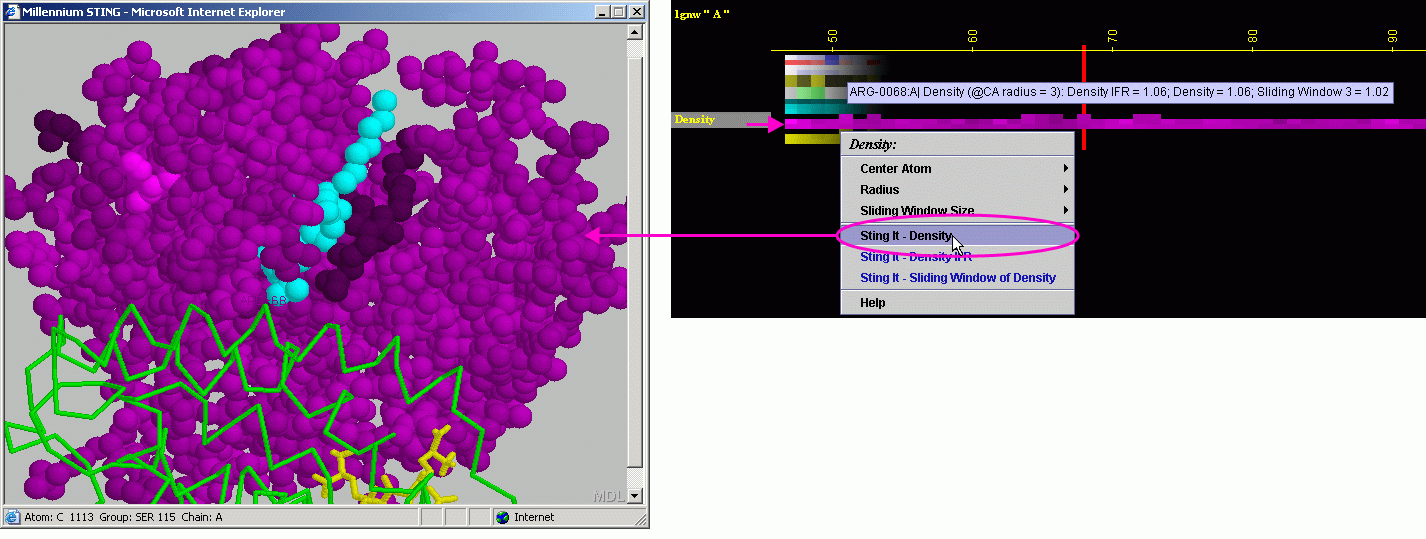
E) STING it "Density
IFR": This option generates structural presentation with color coding
of the amino acids corresponding to the JPD row "Density IFR."
color coding. The area of interface between the two chains is where the
colors are going to be different from the rest of the protein. Amino acids
are presented in CPK rendering.

F) STING it "Sliding
Window Density IFR.": This option generates structural presentation
with color coding of the amino acids corresponding to the JPD row "Density
IFR " color coding. The area of gaps is where the colors are going
to be different from the rest of the protein. Amino acids are presented
in CPK rendering.